
Secondary structure analysis using STRIED in VMD
STRIDE is a widely used secondary structure assignment tool for protein structures. This algorithm was first reported in:
Frishman D, Argos P. Knowledge-Based Protein Secondary Structure Assignment Proteins: Structure, Function, and Genetics 23:566-579 (1995)
Please also check Secondary structure analysis using DSSP.
1. For starters
Generally speaking, protein structures have helix, $\beta$-sheet and coils as their secondary structure elements. The classification derives from the backboe dihedrals.
Backbone dihedrals and Ramachandran plot:
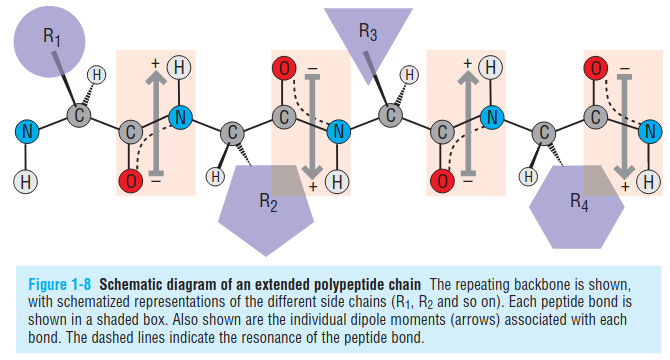
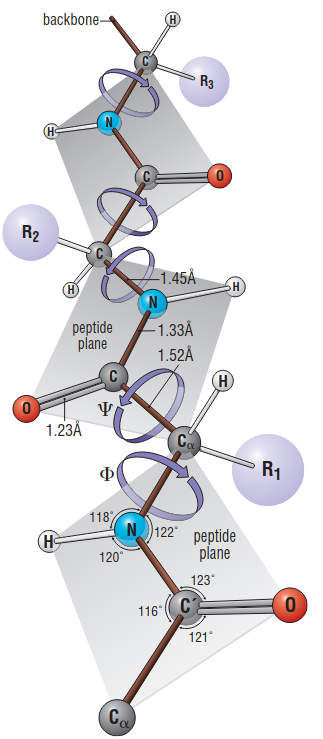
The plenary peptide bond can rotate around $N-C\alpha$ bonds (referred as the phi or $\phi$ dihedral angle) and also $C\alpha-C$ bonds (referred as the psi or $\psi$ dihedral angle).
Statistically, to avoid steric clashes, protein backbone Ramachandran plot (when plotting the $\phi$ and $\psi$ dihedrals of residues as the x and y-axis) should be like the following:
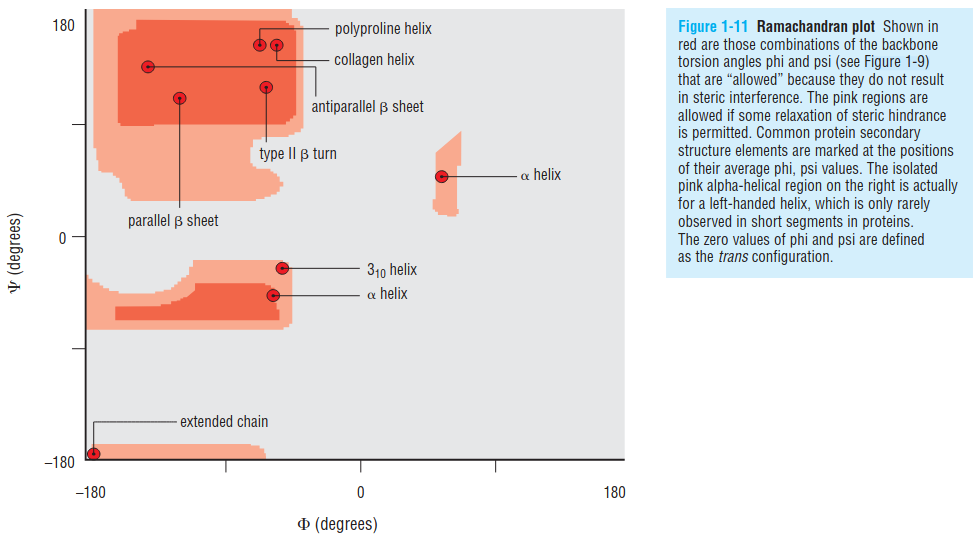
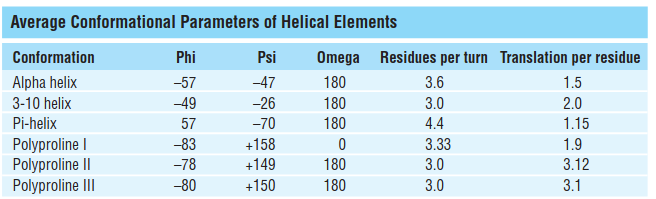
1.1 helix
- In an alpha helix, the carbonyl oxygen atom of each residue (n) accepts a hydrogen bond from the amide nitrogen four residues further along (n+4) in the sequence
- The alpha helix is a compact structure, with approximate phi, psi values of –60° and –50° respectively: the distance between successive residues along the helical axis (translational rise) is only 1.5 Å
- In an alpha helix the hydrogen-bonding pattern causes all of the amides—and their dipole moments—to point in the same direction, roughly parallel to the helical axis
- It would take a helix 20 residues long to span a distance of 30 Å, the thickness of the hydrophobic portion of a lipid bilayer
- Alpha helices can be right-handed (clockwise spiral staircase) or left-handed (counterclockwise), but because all amino acids except glycine in proteins have the L-configuration, steric constraints favor the right-handed helix, as the Ramachandran plot indicates
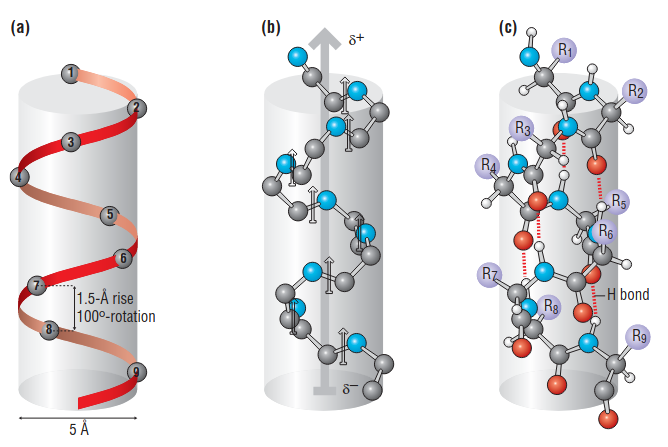
However, it is worth noticing that there are some variations on the helical structures:
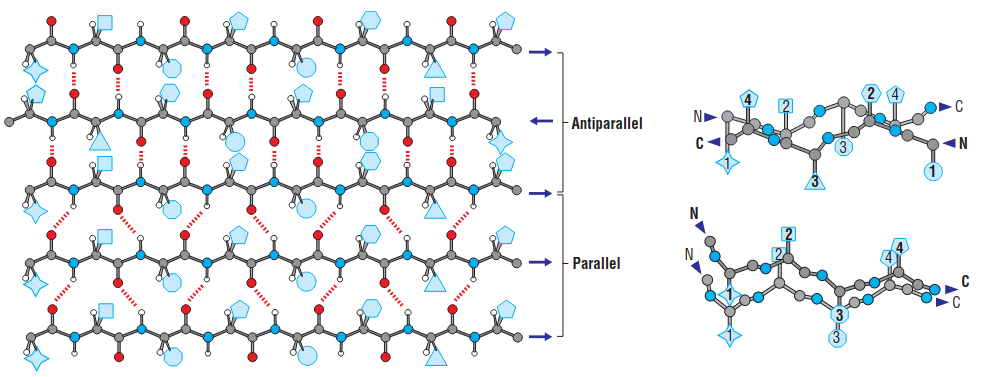
1.2 sheet
- Parallel sheets are always buried and small parallel sheets almost never occur.
- Antiparallel sheets by contrast are frequently exposed to the aqueous environment on one face.
- antiparallel sheets are more stable, which is consistent with their hydrogen bonds being more linear.
- The polypeptide chains that comprise antiparallel pleated sheets tend to have alternating hydrophilic and hydrophobic residues, so that hydrophobic side chains tend to be present on one side of the sheet and hydrophilic residues on the other.
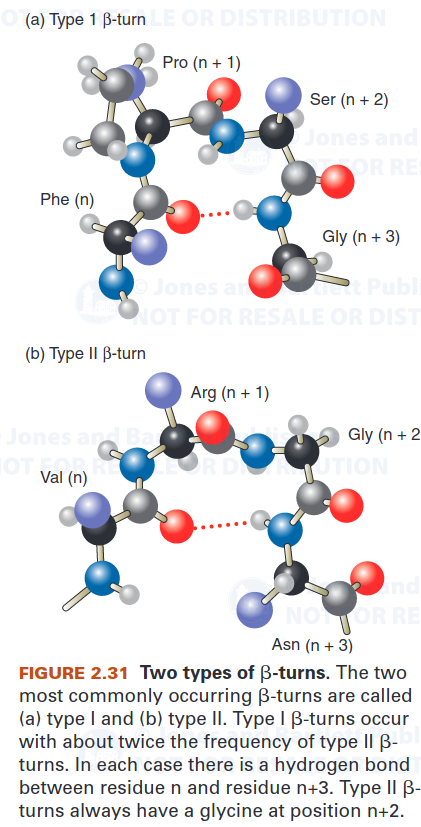
1.3 $\beta$-turn
The most common kind of turn, the $\beta$-turn, consists of four residues and allows the polypeptide chain to reverse direction. The carbonyl oxygen of the first residue in a $\beta$-turn forms a hydrogen bond with the amino group of the fourth residue.
Glycine and proline are commonly present in $\beta$-turns. Glycine’s conformational flexibility allows it to fit into the tight turn. Proline’s steric constraints are also well suited to the $\beta$-turn.
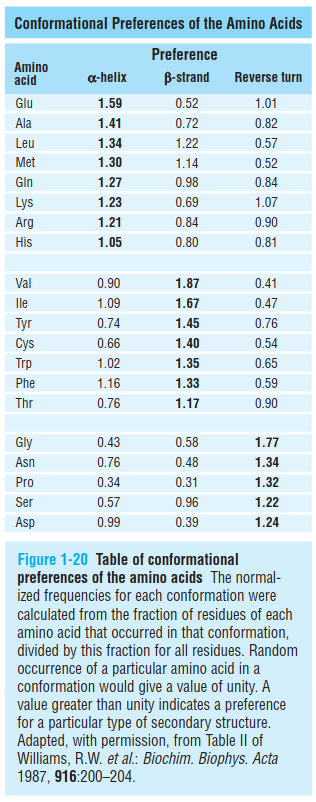
1.4 conformational preferences of amino acids
2. The STRIDE algorithm
STRIDE uses 3D coordinates from PDB files or MD snapshots, like DSSP. It looks at backbone atoms (N, Cα, C, O) as well as side-chain Cβ atoms for additional geometrical information. Like DSSP, STRIDE identifies backbone hydrogen bonds. But STRIDE uses a different energy function that accounts for bond distances and angles more explicitly. Hydrogen bonds are scored energetically, not just via a cutoff threshold, making it slightly more sensitive to subtle H-bond patterns.
STRIDE heavily considers φ (phi) and ψ (psi) backbone dihedral angles. It uses probability distributions of these angles derived from high-resolution crystal structures for each type of secondary structure. Example: α-helix residues cluster in a particular φ/ψ region; β-strands cluster in another region. (see the Ramachandran plot)
STRIDE assigns secondary structure based on a combined scoring function:
\[S_{total} = S_{H-bond} + S_{torsion}\]- $S_{H-bond}$ = score based on hydrogen bond energies.
- $S_{torsion}$ = score based on φ/ψ probability from known structures.
- The residue is assigned the secondary structure maximizing the total score.
STRIDE assigns the same 8 standard types as DSSP (H, G, I, E, B, T, S, C). Because it incorporates torsion angles and probabilities, STRIDE tends to be slightly more sensitive to subtle helices or turns than DSSP. It also tends to give smoother assignments across residues (less “flickering” of assignments in short helices or strands).
3. Key differences from DSSP
| Feature | DSSP | STRIDE |
|---|---|---|
| Hydrogen bond detection | Electrostatic model with energy cutoff | Energy-based scoring with refined geometry |
| Torsion angles | Minor influence | Major component (φ/ψ probability) |
| Side chain atoms | Ignored | Cβ atoms used for geometry |
| Sensitivity | May miss short or distorted helices | Captures subtle helices and bends better |
| Output | 8-letter or 3-state simplified | 8-letter or 3-state simplified |
4. use STRIDE in MD trajectory
VMD has the TimeLine plugin for analyzing secondary structures via STRIDE.
Besides, check the vmd_stride.tcl in my github repo link.
5. Comparison with DSSP
Below is an example of me using DSSP and STRIDE for a series of MD trajectory with a focus on the residue index 205:220.
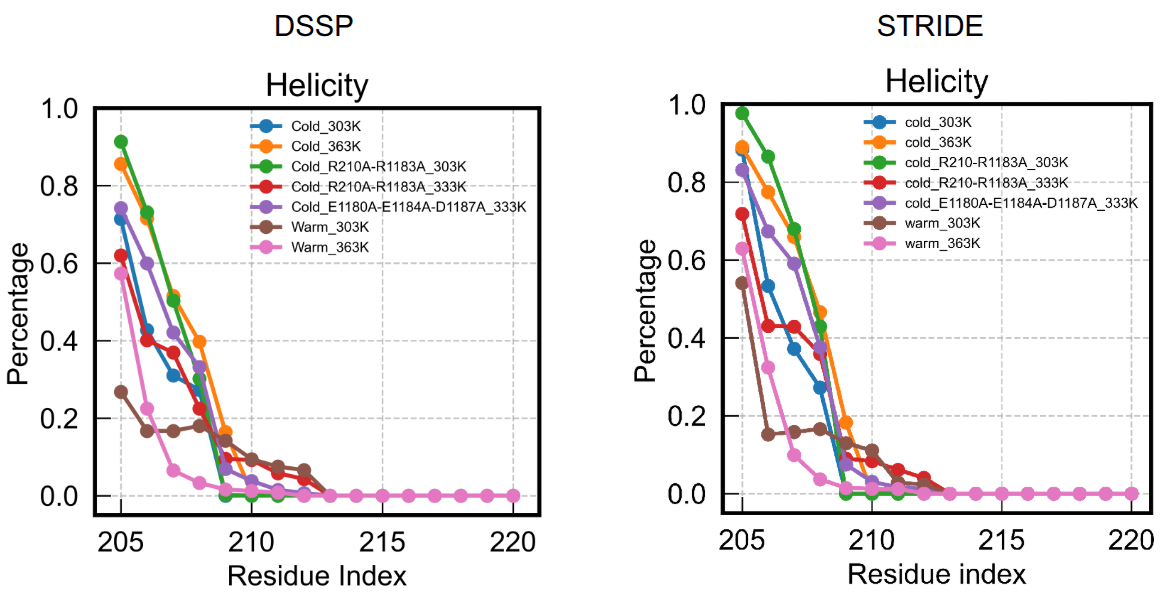
The results largely look similar, though STRIDE overall emphasizes helicity a little bit.