
TRPV4: Gating Mechanisms and Lipid Regulation
TRPV4 is a Ca<sup>2+</sup>-permeable nonselective cation channel with polymodal gating — it can be activated by temperature, osmotic stress, mechanical force, and chemical ligands. Despite the availability of both open- and closed-state structures, the molecular mechanisms underlying its activation and regulation remained poorly understood. Our work addresses two central questions: How does TRPV4 gate ion flow in the closed state? and How does the lipid PIP2 regulate channel activity?
Study 1 — Hydrophobic Gating Co-exists with Bundle Crossing in TRPV4
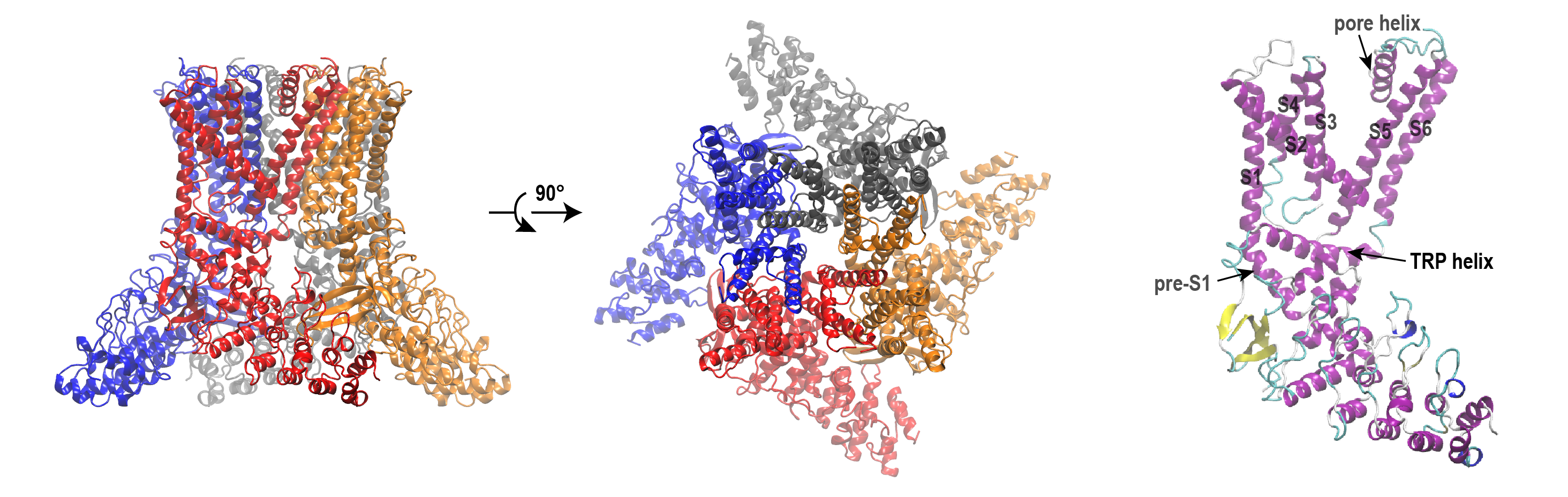
[Huang & Chen, Commun. Biol. 6, 1094 (2023). DOI]
Transmembrane ion channels are classically thought to close by forming a bundle crossing — a physical constriction where the pore-lining helices pack tightly together at the intracellular end. In the closed state of TRPV4, such a bundle crossing is evident. However, the inner pore enclosed by the bundle crossing is lined by highly hydrophobic residues, raising the question of whether physical constriction alone is sufficient to block ion flow.
Using all-atom molecular dynamics (MD) simulations, we demonstrate that TRPV4 deploys a second, complementary gating mechanism — hydrophobic gating — in which the hydrophobic inner pore spontaneously dewets, forming a vapor-phase barrier that blocks ion permeation even in the absence of physical constriction. Key findings:
- Two mechanisms co-exist: bundle-crossing constriction and hydrophobic dewetting both contribute to preventing ion flow in the closed state.
- A single mutation reveals the hydrophobic gate: introducing a hydrophilic substitution I715N near the bundle crossing dramatically increased pore hydration and reduced the free energy barrier for K
<sup>+</sup>permeation by approximately 50%, without any measurable effect on the physical geometry of the bundle crossing. This single-point mutation was previously shown experimentally to increase resting channel activity. - General implication: given the prevalence of hydrophobic inner pores across the ion channel superfamily, hydrophobic gating is likely a more general and underappreciated gating mechanism than previously recognized — even in channels where bundle crossing is considered the primary gate.
Study 2 — PIP2 Binding Sites and Dynamic Domain Coupling in TRPV4
[Huang & Chen, Biophys. J. 124, 3037–3048 (2025). DOI]
Phosphatidylinositol 4,5-bisphosphate (PIP2) is a signaling lipid that modulates many ion channels, including TRPV4. Prior proposals placed PIP2 binding on the N-terminal domain or the ankyrin repeat domain (ARD), but these sites are too distal from the membrane interface to plausibly influence channel gating. We revisited this question using cryo-EM structural analysis, molecular docking, and extensive atomistic MD simulations combined with free energy calculations and dynamic network analysis.
Key findings:
- Two binding sites near the membrane interface: structural analysis and docking revealed two candidate PIP2 binding sites located close to the cytoplasmic membrane surface — a more mechanistically plausible location to influence gating.
- A single broad binding groove: the two sites are not independent; they form a continuous, broad groove in which PIP2 binding is highly dynamic, sampling multiple configurations of interactions with positively charged side chains. The binding affinity is approximately 4 kcal/mol relative to the bulk membrane.
- Allosteric modulation of domain coupling: dynamic network analysis shows that PIP2 occupancy in this groove alters the dynamic coupling between multiple domains of TRPV4 — the transmembrane domain, the coupling domain, and the cytosolic ARD. This suggests a mechanism through which PIP2 could prime or modulate TRPV4’s ability to respond to diverse stimuli including temperature, osmolarity, and mechanical force.
Significance
Together, these two studies provide a more complete picture of TRPV4 gating and regulation at the molecular level. The identification of hydrophobic gating challenges the view that bundle crossing alone governs channel closure, while the PIP2 work establishes a structurally plausible allosteric mechanism for lipid-mediated modulation. Both insights have implications for understanding TRPV4-related channelopathies and for rational drug design targeting this physiologically important channel.