
Dynamic Domain Interactions Encode CheA Autophosphorylation Mechanisms
CheA is the central histidine kinase of the bacterial two-component chemotaxis signaling system. Upon activation by the chemoreceptor–CheW–CheA ternary complex, CheA autophosphorylates the conserved His48 on its P1 (phosphorylatable) domain. This phosphoryl group is subsequently transferred to the response regulator CheY, ultimately controlling flagellar rotation and swimming behavior. Despite decades of structural and biochemical study, the molecular mechanism by which the flexible P1 domain productively engages the kinase P4 domain — and how this is regulated by ATP binding and P1/P1′ dimerization — has remained poorly understood.
[Huang, Wahlbeck Lu-Diaz, Thompson & Chen, Biophys. J. 125, 1125–1138 (2026). DOI]
CheA Architecture and the Autophosphorylation Problem
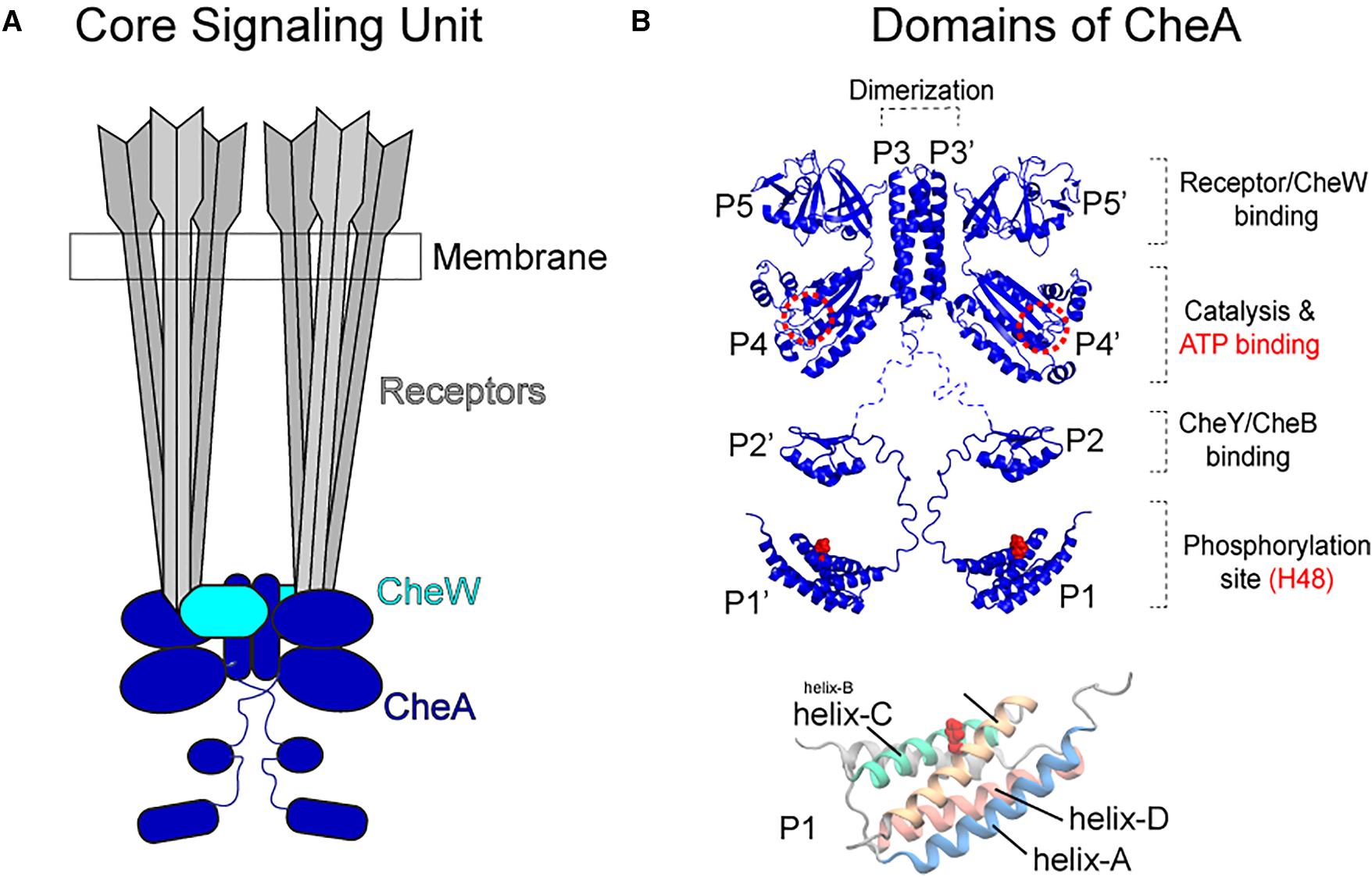
(Left) Topology of the core signaling unit: two CheA dimers, two CheW coupling proteins, and two trimers of chemoreceptors. (Right) Domain organization of a CheA monomer: P1 (substrate, carries His48), P2 (CheY/CheB docking), P3 (dimerization), P4 (ATP-binding kinase), P5 (regulatory, binds CheW and receptors). P1 is connected to the rest of the protein by a long, flexible linker.*
CheA is a homodimer anchored through its P3 domain. The kinase active site resides in P4, while the phosphorylatable His48 is on P1 — tethered to P2 by a disordered linker. Because His48 must reach the ATP γ-phosphate bound in the P4 active site for autophosphorylation to occur, the reaction requires a productive P1–P4 encounter. Both cis (same chain) and trans (opposite chain) P1–P4 contacts are geometrically possible within the dimer. Existing NMR studies, mutagensis as well as chemical modification-based mapping have captured several P1–P4 interaction modes, but it is unclear which represent productive, on-pathway encounters versus nonproductive dead ends — and how ATP binding or P1/P1′ dimerization modulates the ensemble.
HyRes Coarse-Grained Simulations of Full-Length CheA
To sample the large-scale, slow P1 domain dynamics that are inaccessible by all-atom MD on tractable timescales, we used the HyRes coarse-grained (CG) force field. HyRes represents each residue by backbone heavy atoms plus a CG side-chain bead, retaining backbone hydrogen-bonding and secondary structure propensities while dramatically accelerating conformational sampling. Full-length CheA dimers were simulated in the presence and absence of ATP (modeled as a modified nucleotide bead) for a total of hundreds of microseconds of CG simulation time.
ATP Binding Promotes P1 Sampling Near P4
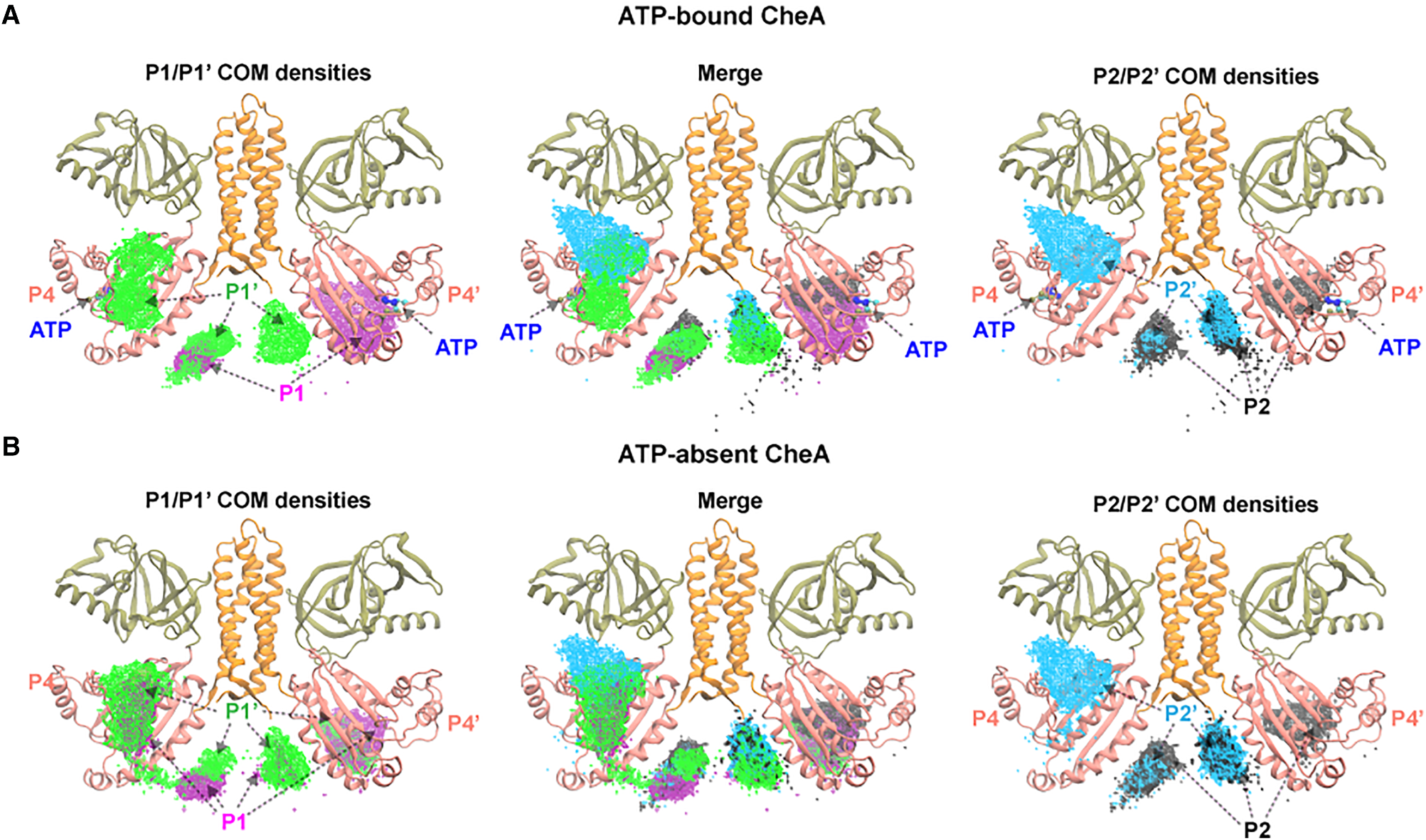
Center-of-mass (COM) density of the P1 domain (both chains) mapped around the CheA dimer in (A) ATP-bound and (B) ATP-absent simulations. White dashed circles indicate the P4 domains.
Without ATP, the P1 domains sample a broad, diffuse region around the dimer, with no strong preference for the P4 kinase domain. Upon ATP binding, P1 density becomes significantly enriched near P4 — both on the same chain (cis) and the partner chain (trans). This redistribution demonstrates that the nucleotide allosterically reshapes the P1 free-energy landscape, funneling the flexible domain toward productive encounter geometries. The trans enrichment is particularly striking and supports the long-standing hypothesis that CheA autophosphorylation proceeds predominantly in trans.
Productive P1–P4 Docking Configurations
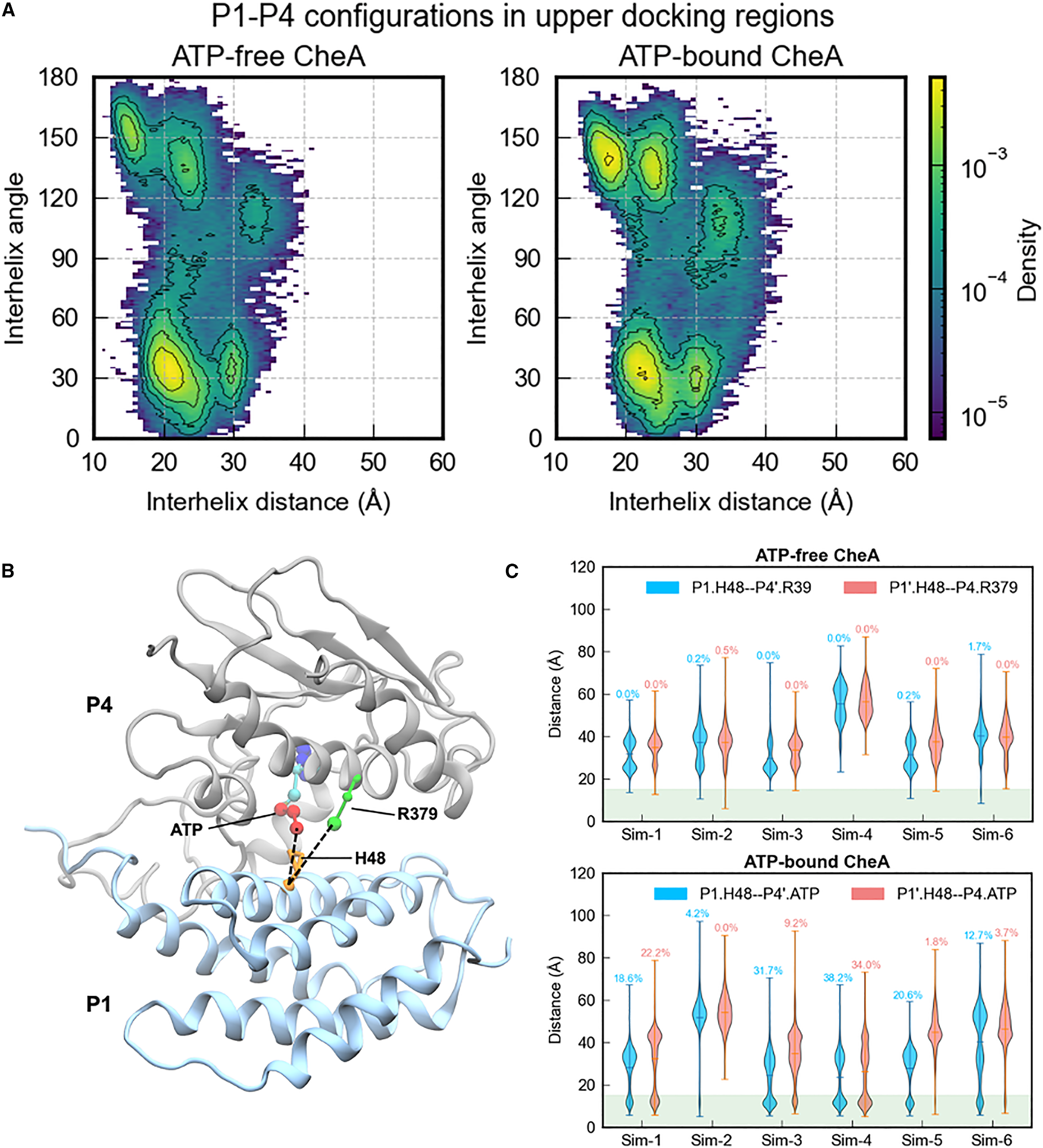
(Top row) Representative P1–P4 upper docking configurations from ATP-bound simulations (P1 in blue, P4 in gray, ATP in orange, His48 in red). (Bottom row) Violin plots of the His48–ATP distance distributions for each configuration, with the ~8 Å cutoff (dashed line) used to define productive contacts.*
We identified and classified P1–P4 docking modes by clustering the P1 orientation relative to P4. Productive contacts — defined as His48 within ~8 Å of the ATP γ-phosphate — were observed in two distinct binding modes.
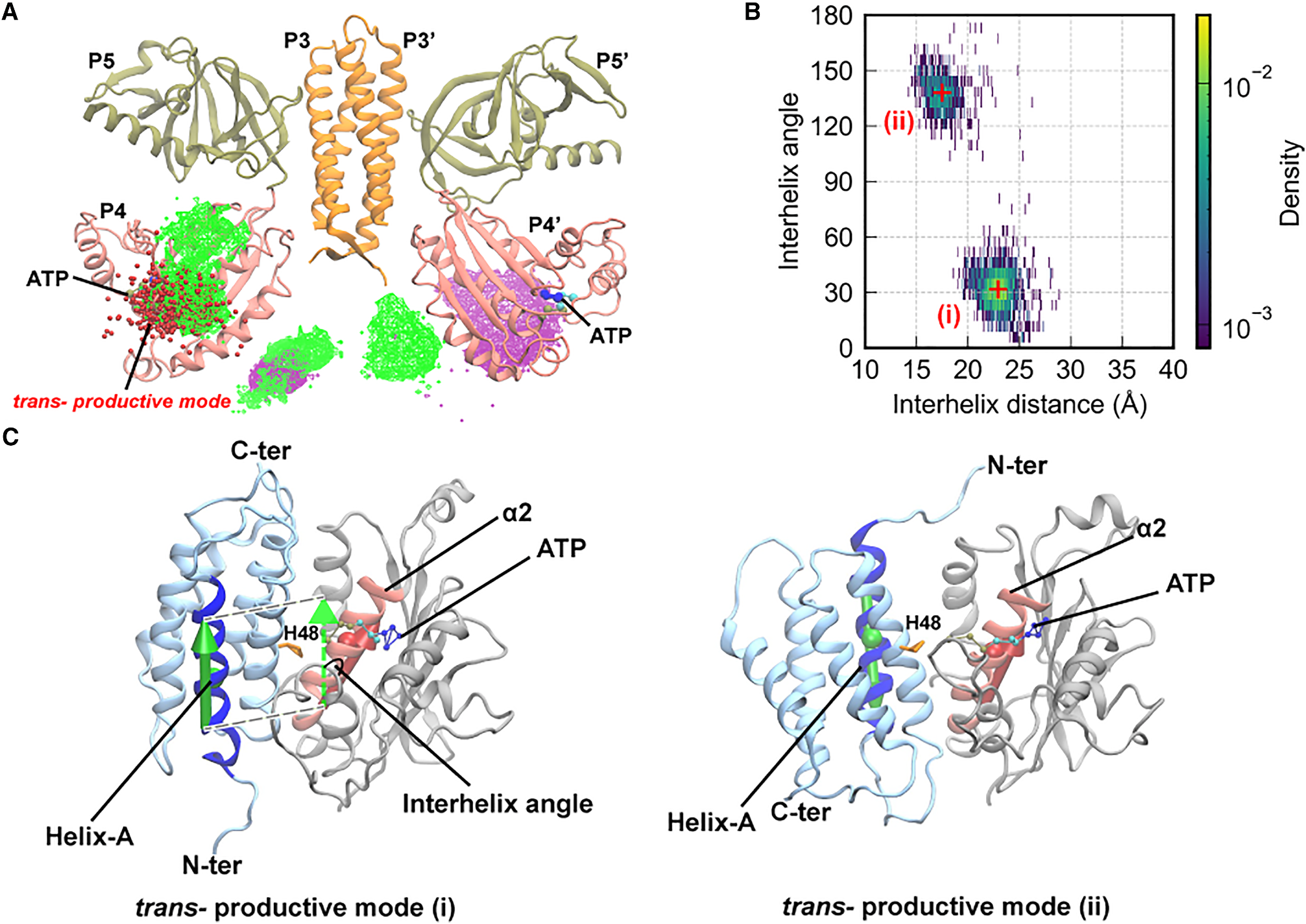
Productive *trans P1/P4 binding modes. (Mode i) P1 approaches from the lateral face of P4, with His48 positioned to attack ATP from the side. (Mode ii) P1 docks onto the top of P4 near the ATP lid, with His48 entering from above. In both modes, His48–ATP distance falls below 8 Å (red distributions).*
Mode (i): P1 engages the lateral surface of P4, with His48 inserting into the ATP-binding cleft from the side. This configuration closely resembles one of the crystallographic P1–P4 complexes and supports a direct in-line phosphoryl transfer geometry.
Mode (ii): P1 approaches P4 from above, with His48 reaching into the active site over the ATP lid. This mode is less frequently sampled but yields similarly short His48–ATP distances, suggesting it is a viable alternative productive pathway.
Both modes are enhanced in the ATP-bound state, consistent with the COM density analysis showing that ATP promotes P1 accumulation near P4.
Nonproductive Binding Modes Sequester P1
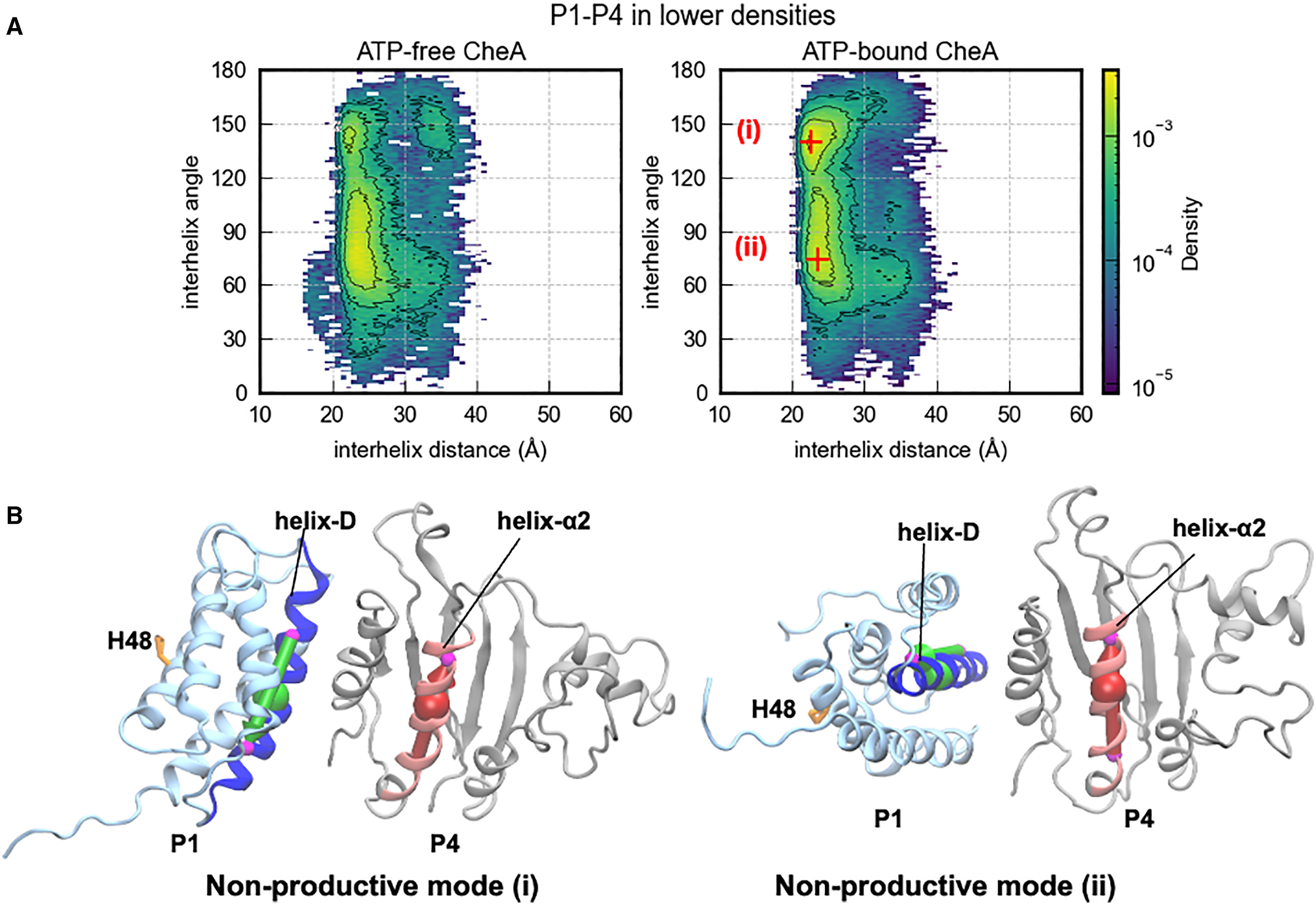
Nonproductive P1/P4 binding modes (i) and (ii), in which P1 is docked on P4 but His48 is oriented away from ATP (His48–ATP > 15 Å in both cases).
In addition to productive contacts, P1 samples two major nonproductive docking modes where it binds stably to P4 but with His48 pointing away from ATP. These modes likely represent kinetically trapped states that must be escaped before productive phosphoryl transfer can occur. Their existence rationalizes why CheA autophosphorylation is relatively slow despite frequent P1–P4 encounters: a significant fraction of binding events are nonproductive.
P1/P1′ Dimerization as an Autoinhibitory Mechanism
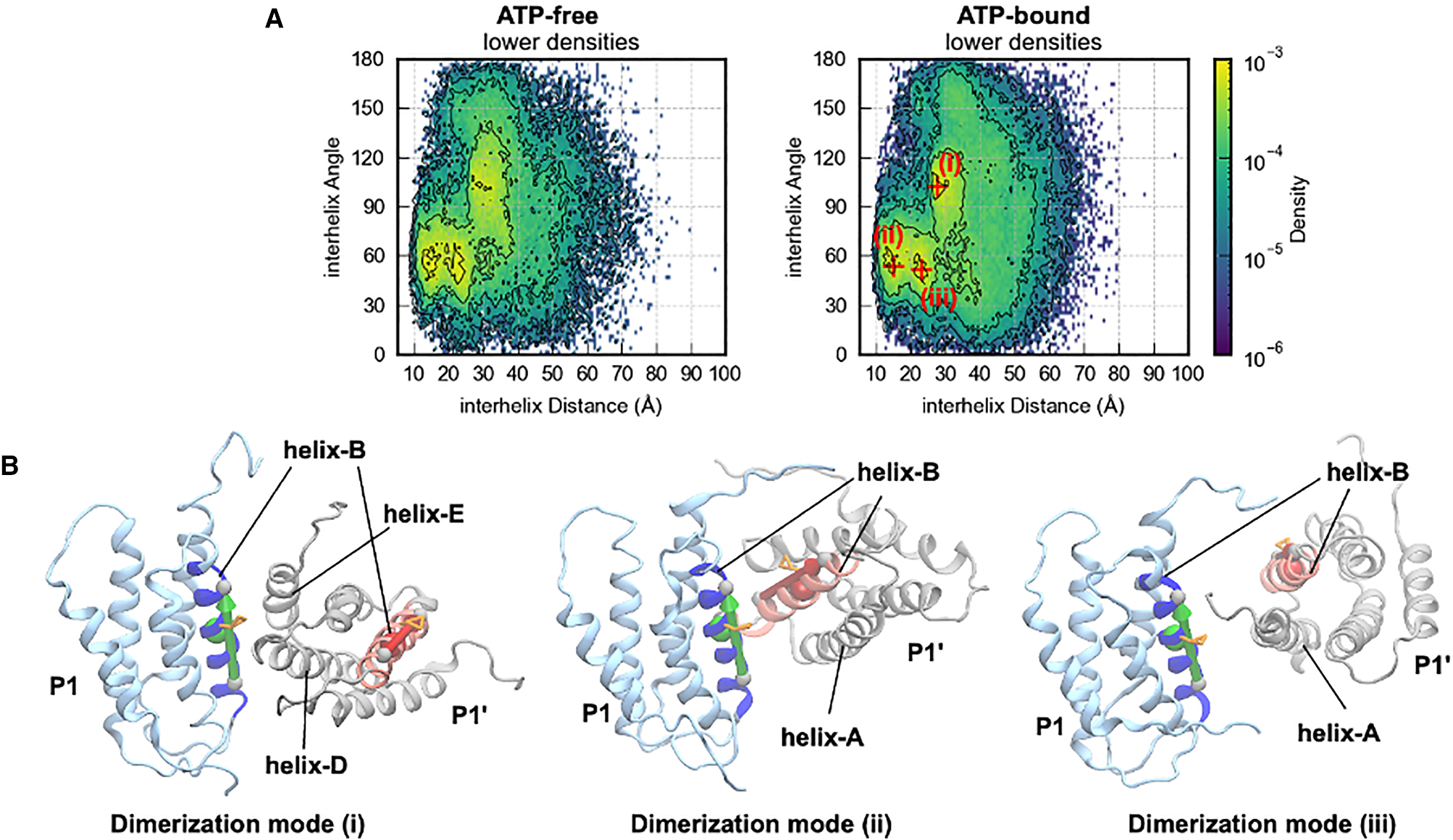
Three P1/P1′ homodimerization modes observed in simulations. Mode (i): symmetric antiparallel bundle. Mode (ii): asymmetric bundle. Mode (iii): extended interface. In all modes, His48 residues of both chains are buried at the dimer interface, rendering them inaccessible to ATP.
The P1 domain is known to dimerize through its helix bundle, but the functional consequences were unclear. Our simulations reveal three distinct P1/P1′ dimerization geometries, each burying His48 at the interface. When P1 is engaged in homodimerization, it cannot simultaneously dock productively onto P4 — providing a direct structural basis for autoinhibition. The equilibrium between P1/P1′ dimerization and P1–P4 productive contacts thus acts as a rheostat: conditions that destabilize P1 homodimerization (such as receptor/CheW activation of CheA) would shift the ensemble toward productive P1–P4 encounters and increase autophosphorylation rate.
Significance
This work provides the first comprehensive dynamic picture of how the phosphorylatable P1 domain navigates its conformational landscape to autophosphorylate CheA. The key contributions are: (1) demonstration that ATP binding allosterically promotes P1 localization near the P4 kinase domain; (2) identification of two distinct productive trans P1–P4 docking geometries that bring His48 within striking distance of ATP; (3) characterization of nonproductive binding modes that compete with productive contacts and slow overall autophosphorylation; and (4) discovery of three P1/P1′ homodimerization modes that sequester His48 and provide a structural basis for autoinhibition. Together, these findings establish a dynamic ensemble model for CheA autophosphorylation regulation and offer a mechanistic framework for understanding how receptor activation modulates kinase activity in bacterial chemotaxis.
Code and simulation data are available at github.com/huangjianhuster/CheA_interdomain_interactions.