
Physics-Informed Deep Learning for Protein Variant Effect Prediction
Predicting how mutations affect protein function — so-called variant effect prediction (VEP) — is a central challenge in protein engineering and the study of genetic disease. Deep learning models trained on deep mutational scanning (DMS) data have shown impressive performance, but they share a critical blind spot: extrapolation. When asked to predict mutations at positions or of mutation types not represented in training data, purely sequence-based models fail badly. This work addresses that limitation by embedding biophysical knowledge directly into the learning pipeline.
[Barethiya, Huang, Stumpf, Liu, Guan & Chen, PLOS Comput. Biol. (2026). DOI]
The Extrapolation Problem
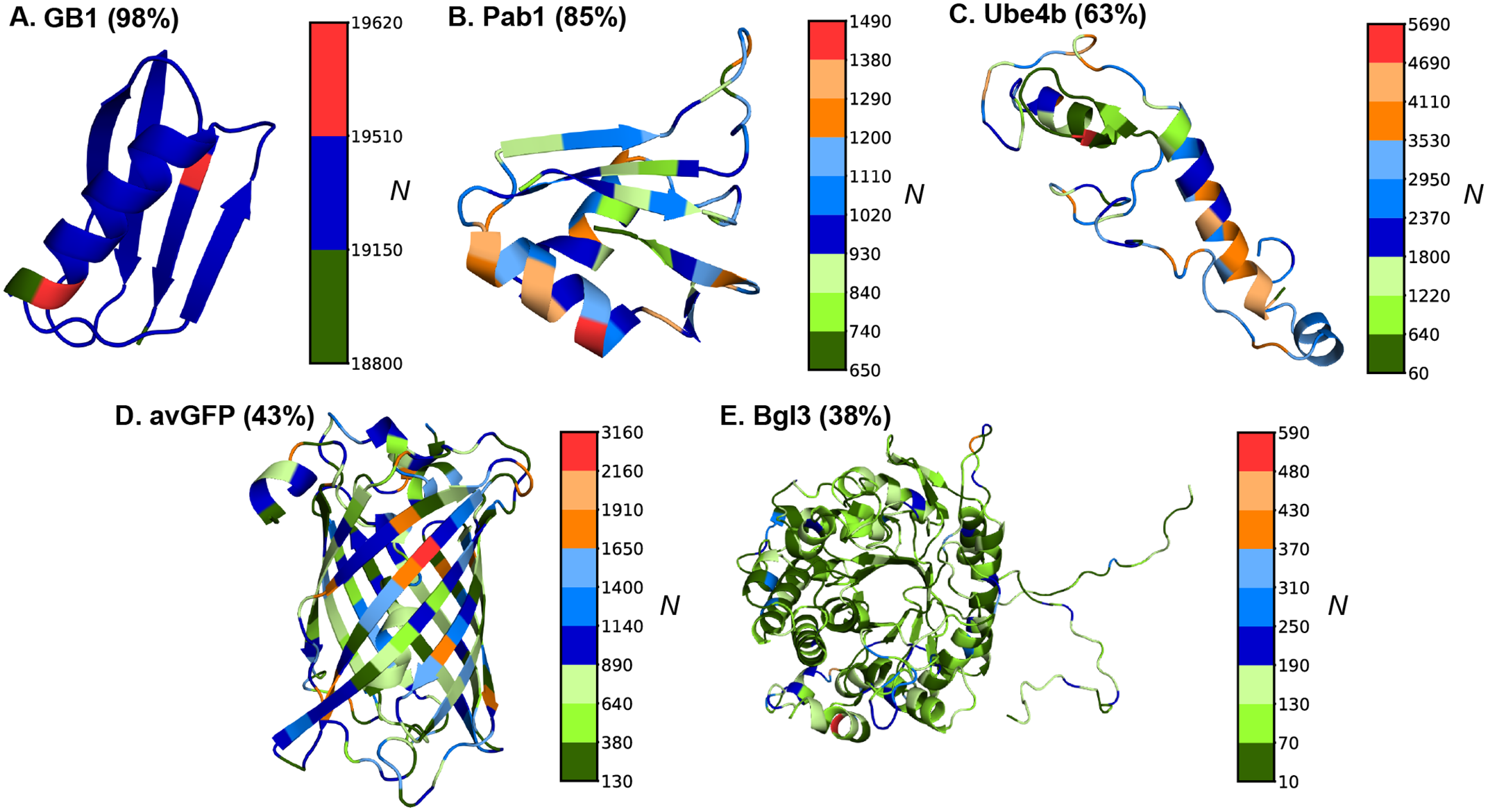
Protein structures and mutation coverage maps for the five benchmark proteins (GB1, Pab1, avGFP, Bgl3, Ube4b) spanning 56–501 residues and 38–98% positional coverage.
Standard deep learning models for VEP interpolate well — they reliably assign fitness scores to mutations similar to ones seen during training. But real-world applications demand more: predicting effects at positions absent from the training library (positional extrapolation), generalizing from single-residue variants to higher-order combinatorial mutations (order extrapolation), and handling novel mutations (mutational extrapolation). Sequence-only models consistently collapse under these conditions, often reaching near-zero correlation with experimental fitness.
A Physics-Informed Framework
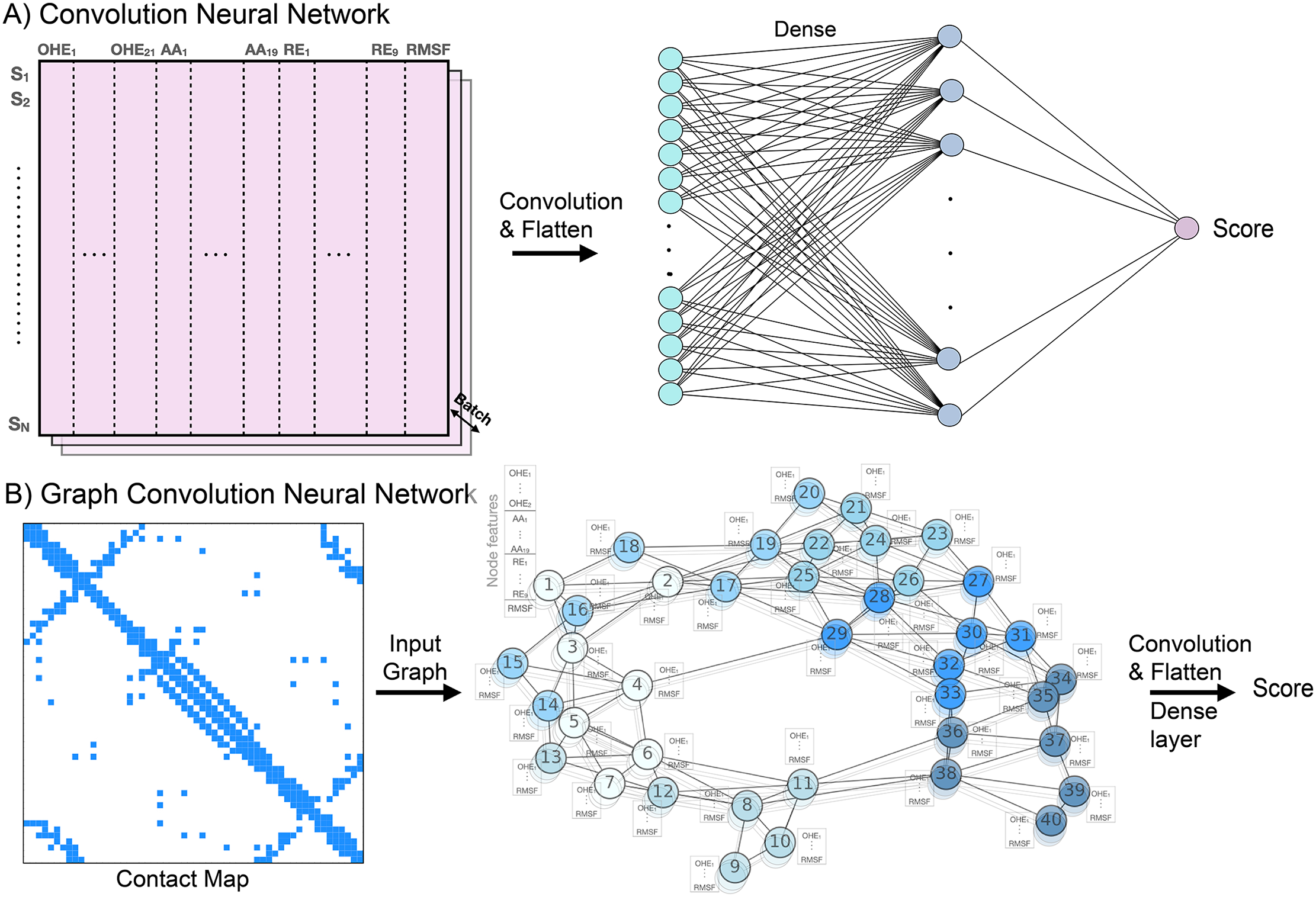
CNN and GCN model architectures showing how biophysical feature channels (Rosetta energy terms + MD-derived RMSF) are incorporated alongside sequence representations.
We augment convolutional neural networks (CNN) and graph convolutional networks (GCN) with two classes of biophysical features computed from protein structure:
- Rosetta energy features: Eight residue-level energy terms (van der Waals, solvation, electrostatics, hydrogen bonding, etc.) calculated via a local rotamer repacking protocol. For each mutation, only the site and its neighbors within a 12 Å radius are repacked — requiring just sequence length × 19 Rosetta evaluations per protein, making this computationally tractable.
- MD-derived flexibility (RMSF): Backbone fluctuation profiles from atomistic MD simulations encode the dynamic context of each residue, capturing information not accessible from static structures alone.
These features are concatenated with sequence embeddings (one-hot or ESM-2 language model representations) and fed into CNN or GCN architectures, which are then trained and tested on five benchmark DMS datasets covering diverse protein folds and functional classes.
Key Results
Under random splitting (standard benchmark), biophysics features provide modest but consistent improvements, most notably when training data is sparse (under 1,000 variants) or when sequencing quality is low — conditions where the model must lean more heavily on physically meaningful priors.
Under mutational extrapolation (training on, e.g., certain subsets of mutations; testing on mutation types not seen):
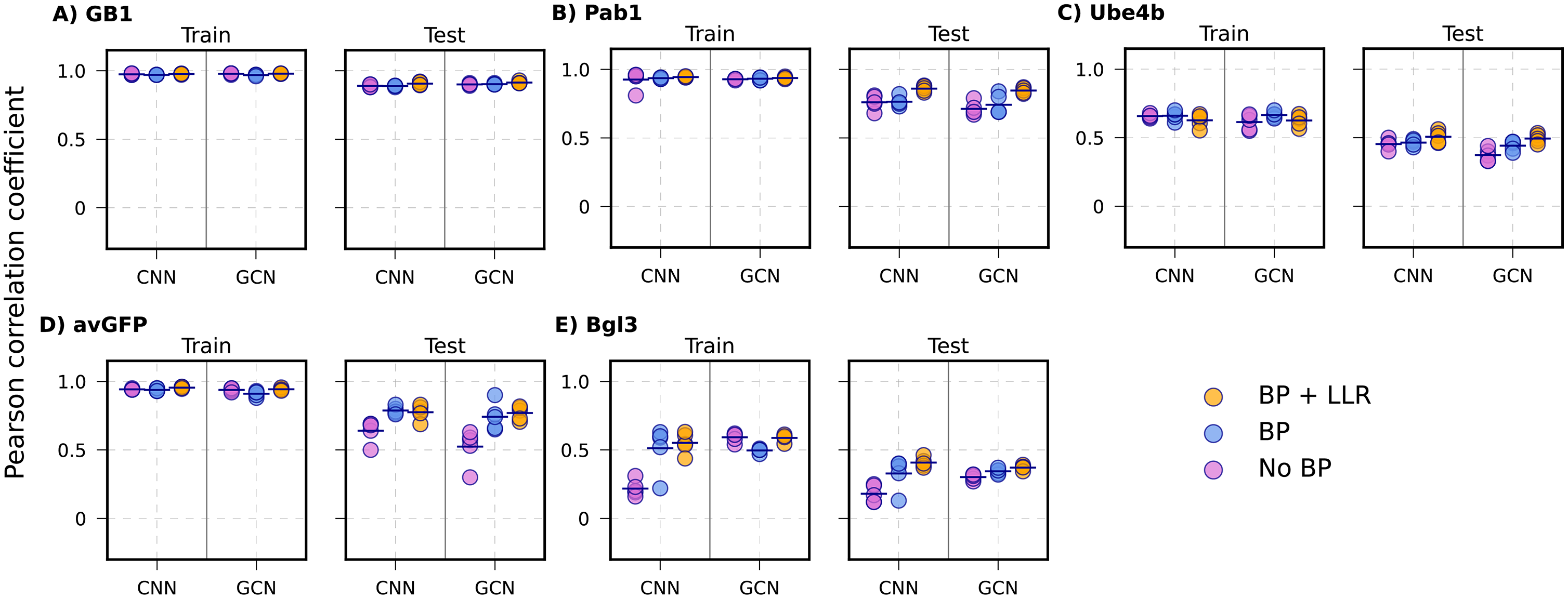
Mutational extrapolation results. Biophysics-augmented models (orange/red) substantially outperform sequence-only baselines (blue) across all five proteins and both CNN/GCN architectures.
The gains are dramatic. For avGFP, a linear model with biophysics improves from r = 0.06 to r = 0.61. CNN/GCN models reach r = 0.52–0.74. The biophysical features provide the model with transferable physical constraints that generalize across mutation types.
Under positional extrapolation (training on certain positions while testing on other positions not trained on):
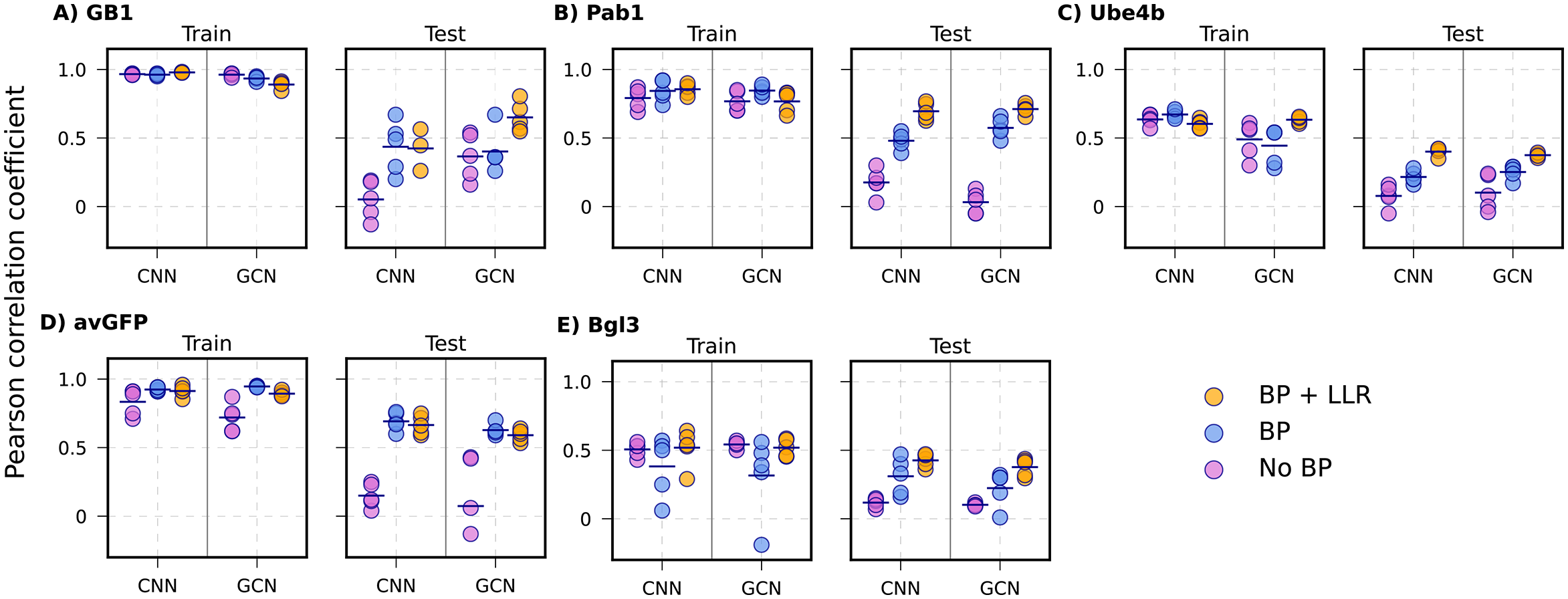
Positional extrapolation results. The most challenging scenario shows the largest improvements — GB1 CNN improves from r = 0.05 to r = 0.43; Pab1 GCN from r = 0.03 to r = 0.57; avGFP from r = 0.15 to r = 0.69.
This is the hardest setting and also where the benefit of physics is most striking. Feature ablation analysis confirms the biophysical terms act synergistically — removing any individual Rosetta energy term substantially degrades performance, indicating the model learns from their combined physical picture rather than any single term in isolation.
Incorporating ESM-2 evolutionary embeddings on top of the biophysical features yields additional consistent gains, showing that evolutionary and physical information are complementary.
Significance
This work establishes that biophysics-based features are a reliable solution to the extrapolation problem in data-driven VEP models. The approach is efficient — it does not require end-to-end structure prediction or large-scale MD for every variant — and broadly applicable to any protein with an experimental or predicted structure. The framework is particularly valuable for protein engineering campaigns where experimental coverage of sequence space is inherently incomplete, and for clinical variant interpretation where mutations of interest are by definition novel.
Code and training data are available at github.com/SBarethiya/VEP.